2022年,长期存在的IVDD将被IVDR取代,开启欧盟市场体外诊断器械监管新纪元。这意味着希望获得 CE 标志并在欧洲销售其产品的制造商将面临一系列新的挑战。今天我们就来看下病毒采样管、实验耗材出口欧盟IVDRCE认证怎么办理?

哪些IVD属于Class A类呢?
IVDR法规共有7条分类规则
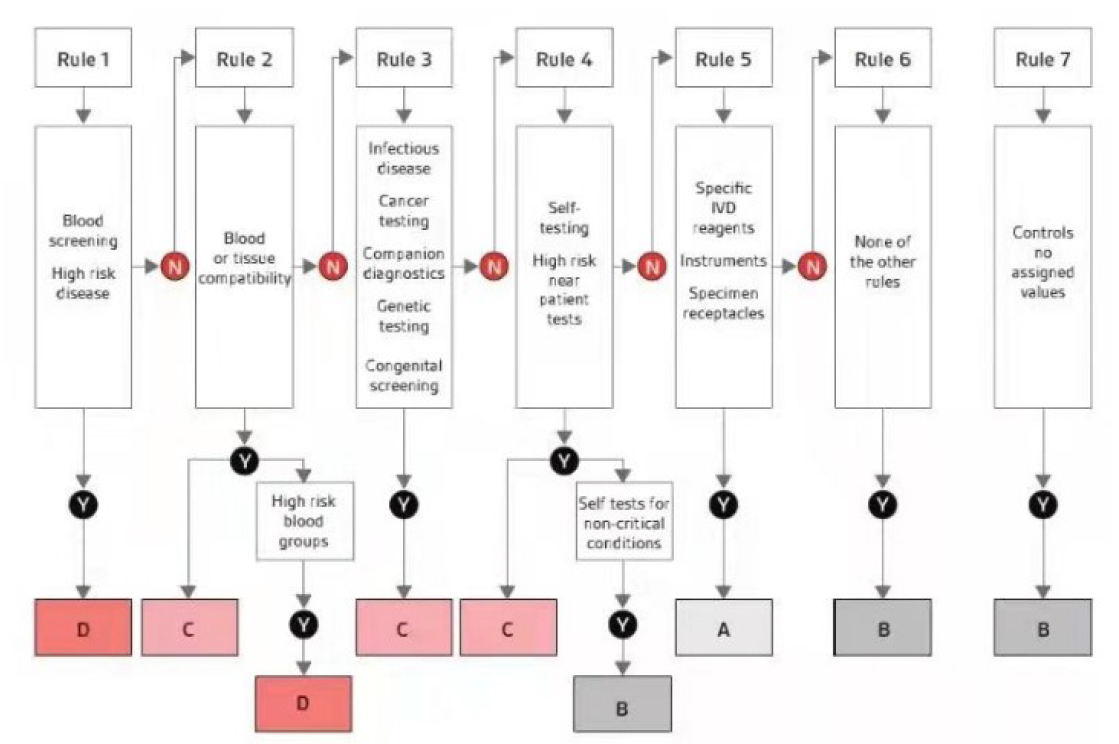
1. Class A类
1)一般实验室使用的产品、不具有关键特性的附件、缓冲液、洗涤液以及一般培养基和组织学染色剂。制造育将此类产品用刊定啮相关的体外诊断。
规则5a适用于一般实验室产品,如移液器、染色粉、显微镜玻璃载玻片、离心机、移液管吸头或仪器液体收集容器、缓冲液。如果制造育专门将此类产品用于体外诊断检查,则它们被视为IVD并适用于规则5。
2)制造商专门用于体外诊断程序的仪器
规则5b适用于制造商专门用于体外诊断程序的仪器。这些仪器被归类为A类,而对应的试剂和试剂盒则根据自身特性进行分类。
3)标本容器
示例(非全部涵盖):真空或非真空管,空的或预装固定液或箕他通用试剂,以保持生物标本的状态,用于运输、储存和收集体外诊断检查的目的。
如果A类IVD产品不是无菌状态交付的产品,那么通过符合性声明程序,欧盟授权代表并完成CE注册就可以加贴CE标志。
同时需要特别说明的是,对于A类IVD产品以无菌状态交付的,需要通过公告机构亩核并获得EC认证证书之后,才能够提交欧盟授权代表完成CE注册。
按照欧盟IVDR法规规定,其技术文件的结构通常需要包括如下内容。当然对于Class A类器械有部分要求不适用,需要在技术文件中进行识别。

2017/746年欧盟(EU)医疗器械体外诊断法规(IVDR)对IVD行业和销售到欧盟市场的公司产生了重大影响,因为他们准备满足更严格的医疗器械合规要求。根据该指令,2022年5月26日之后上市的所有类别的产品必须重新评估是否符合IVDR要求,以确保这些产品适合用途且使用安全。没有基于产品上市时间授予的自动通行证,不允许继续使用。
- MDR认证
- 暂无标签



