IVDR CE认证是指符合欧洲联盟体外诊断器械法规(In Vitro Diagnostic Medical Device Regulation,IVDR)要求的体外诊断器械产品获得CE标志的认证过程。
随着欧盟监管当局2021年12月21日发布公告,IVDR法规实施日期2022年5月26日起,制造商需要在此日期之后需要完成CE技术文件和产品欧盟注册,才能继续在欧盟市场销售其产品。
医疗器械CE认证之体外诊断医疗器械IVDR分类:
IVDR 2017/746号法规附录VII中详定7条规则,按医疗产品的风险程度,将产品分为Class A、Class B、Class C和Class D。
根据这种情况,越来越多的医疗器械制造商开始根据新政策重新考虑销往欧洲的体外诊断产品布局。
由于IVDR法规对体外诊断类产品进行了重新分类,例:新冠RT-PCR试剂在IVDR法规下被归类为D类风险等级产品,在新规执行之后上市就非常有难度。
IVDR法规下,分类为风险更高的类别;那整个费用投入都会大大增加,不仅涉及到公告机构的费用,可能还需要额外增加临床试验费用。
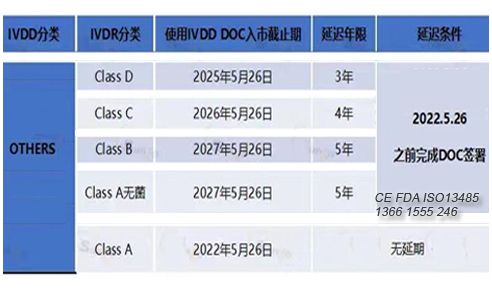
对于IVDD分类为others类的体外诊断医疗器械,其过渡期及合规上市的时间如下:
ClassA 类
1)一般实验室使用的产品、不具有关键特性的附件、缓冲液、洗涤液以及一般培养基和组织学染色剂。制造商将此类产品用于特定检查相关的体外诊断。
规则5a适用于一般实验室产品,如移液器、染色粉、显微镜玻璃载玻片、离心机、移液管吸头或仪器液体收集容器、缓冲液。如果制造商专门将此类产品用于体外诊断检查,则它们被视为IVD并适用于规则5。
2)制造商专门用于体外诊断程序的仪器规则5b适用于制造商专门用于体外诊断程序的仪器。这些仪器被归类为A类,而对应的试剂和试剂盒则根据自身特性进行分类。
3)标本容器
示例(非全部涵盖):真空或非真空管,空的或预装固定液或其他通用试剂,以保持生物标本的状态,用于运输、储存和收集体外诊断检查的目的。

如果A类IVD产品不是无菌状态交付的产品,那么通过符合性声明程序,指定欧盟授权代表并完成CE注册就可以加贴CE标志。
同时需要特别说明的是,对于A类IVD产品以无菌状态交付的,需要通过公告机构审核并获得CE认证证书之后,才能够提交欧盟授权代表完成CE注册。
FDASUNGO荷兰公司已经为国内器械厂商成功申请IVDR CE注册证书。同时,FDASUNGO德国也已经受理部分厂商的IVDR CE注册申请。除了提供欧盟授权代表和注册服务,我们还提供IVDR CE技术文件编撰、IVDR体系升级服务,帮助制造商全面规避合规风险。
- MDR认证
- 暂无标签



