2017年欧盟官方期刊正式发布了欧盟医疗器械法规(Regulation (EU) 2017 / 745,简称“MDR”)。 MDR于2017年5月26日正式生效,在经过3年的过渡期后,将于2020年5月26日正式取代原有的MDD医疗器械(93 / 42 / EEC)和AIMDD有源植入式医疗器械(90 / 385 / EEC)指令。体外诊断试剂法规IVDR也将于2022年5月26日全面执行。
本文重点分析MDR,但有关过渡期的举例同样适用于IVDR。
MDR第120条概括了过渡期的内容,但是很多地方说得比较模糊,令人费解。在此我们主要分享一些相对比较重要的内容,希望能给打算或正在进入欧洲市场的医疗器械从业者们提供参考。
1. CE证书有效时间
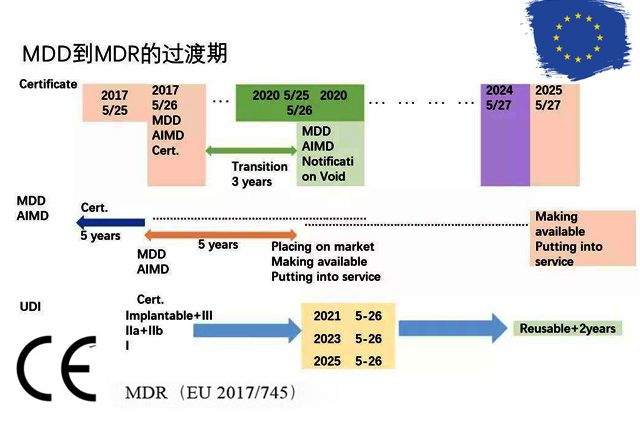
2017年5月25日之前拿到的CE证书,其有效时间将继续保持。
2. CE证书的延续
从今年夏天开始,认证机构(如TUV)会陆续停止按照旧MDD发放CE证书。目前只有两家被允许按照新MDR审理的公告机构,BSI 和 TUV南德。
所以建议2015年5月26日之前拿到CE证书的制造商,应尽快联系这两家认证机构,来按照新法规取得CE证书,以免因新规运行的不确定性因素延误证书的下发时间。
3. 旧证作废时间
按照新规要求,截止2024年5月27日,依据旧指令MDD颁发的CE证书将全部作废。实际操作中,认证机构将不会批准超过此日期的证书(如02条所指,今年夏天认证机构已经陆续停止颁发旧证),因为超过此认证日期的医疗器械将不允许投入市场。
但需要注意的是,制造商仍旧要对已经投入市场并且正在使用的医疗器械负责,继续履行市场不良事件报告的义务。
4. 入市周转时间
如果医疗器械只是进入市场,仍在经销商手中,没有到达最终消费者,那么这种情况下市场的有效运转期将推迟到2025年5月27日。
举例说明:
◆如果你的医疗器械是I类*(有测量功能或无菌医疗器械)或者高于II类(IIa, IIb, III),CE证书是在2018年7月1日颁发,鉴于CE证书的有效期一般是5年,所以在2023年7月1日前制造商仍可以把产品投入欧洲市场。如果已经进入市场,那么可以在2025年5月27日前运转到最终消费者。
◆如果你的医疗器械是I类*(有测量功能或无菌医疗器械)或者高于II类(IIa, IIb, III),而CE证书是在2019年7月1日颁发,那么2024年5月27日前制造商仍可以把产品投入市场(如03条指出,2024年5月27日旧规CE证书全部废止)。如果产品已经进入市场,那么可以在2025年5月27日前运转到最终消费者。
◆如果你的医疗器械是I类,因为无需认证机构参与,制造商自行签署符合性声明(等效于CE证书),无论符合性声明时间如何,过渡期均采用一刀切进行处理,在2020年5月25日之前制造商可以把产品投入市场。如果已经进入市场,那么可以在2025年5月27日前运转到最终消费者。
或者您可以联系FDASUNGO,CE认证、MDR合规咨询,MDR文件编写,第四版临床评估报告编写,欧代服务等。

洞悉MDR新规和过渡期操作,对于医疗器械从业者至关重要。
比如,对于已经获得CE认证的制造商,应尽快把产品交给进口商、经销商和批发商,这样最起码可以保证2025年5月27日前产品仍可运转给最终消费者。
而对于进口商、经销商和批发商,如果对MDR的理解意识超前,那么在2020年新规生效之前,不会轻易接收制造商的产品,因为其无法准确知悉新规需要履行的义务。
- MDR认证
- 暂无标签



