2020年医疗器械CE认证行业,新冠体外诊断试剂的CE认证是最热事项之一。从目前情况看,疫情可能还将持续一段时间,因此,带大家一起来回顾新冠体外诊断试剂CE认证流程和要求。
考虑到IVDD及IVDR对体外诊断试剂的分类存在较大差异,IVDR正式实施后,企业申请医疗器械CE认证,能够豁免临床实验的诊断试剂范围将缩小,因此,对于有志于开拓欧盟市场的体外诊断试剂的客户来说,现阶段是申请医疗器械CE认证的好机会。
为了解决国外检测试剂紧缺局面,截至目前,多家国内体外诊断企业宣布其新冠病毒检测试剂已经获得了欧盟CE准入市场资格,进入了欧盟国际市场。
什么是CE认证呢?
CE代表是欧洲统一(CONFORMITE EUROPEENNE).在欧盟市场“CE"标志属于强制性认证标志,要在欧盟市场上自由流通,就必须加贴"CE"标志。
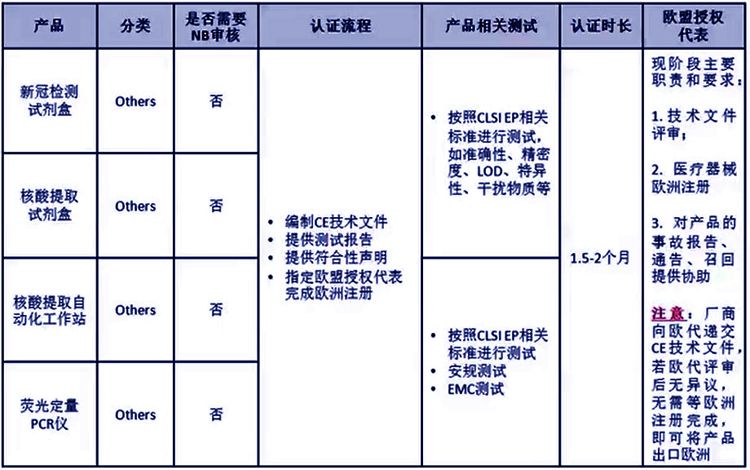
新冠核酸检测试剂盒属于Others类别的,我司服务体外诊断试剂公司有很多,可提供新型冠状病毒核酸检测试剂盒的IVD CE技术文件编写、欧盟授权代表、欧盟注册(MHRA注册、CIBG注册)、欧盟自由销售证书FSC。
体外诊断产品(IVDD)认证知识介绍:
在医疗器械领域,欧盟委员会制定了三个欧盟指令,以替代原来各成员的认可体系,使有关这类产品投放市场的规定协调一致。指令分别是:主动植入式医疗器材 (AIMD 90/385/EEC)/ 医疗器材 (MDD 93/42/EEC) / 体外诊断医疗器材 (IVDD 98/79/EC)。
我国医疗器械生产企业及出口公司在将其产品销往欧盟市场时,必须符合上述指令规定,加贴CE标志,否则产品难以进入欧盟市场。由于我国在这方面宣传力度不够,虽然上述三个指令中的AIMD已强制实施多年,IVD也于1999年强制实施,但目前还有众多的医疗器械生产厂家及出口公司对这些要求不甚了解,因此今年我国的医疗器械出口将面临严峻的考验。
体外诊断CE认证:
1:关于体外诊断医疗器械 (IVDD 98/79/EC)指令:
98/79/EC体外诊断医疗器材指令(简称IVDD指令),适用于血细胞计数器,妊娠检测装置等活体外诊断用医疗器械。该指令已于2000年6月7日生效,将于2005年12月7日强制实施。指令正式实施后,只有带有CE标志的体外诊断医疗器材产品才能在欧盟市场上销售。
2:体外诊断医疗器械的定义:
“体外诊断医疗器械”是指制造商预定用于体外检查从人体取得的样品,包括血液及组织供体的,无论单独使用或是组合使用的任何医疗器械,包括试剂、试剂产品、校准材料、控制材料、成套工具、仪表、装置、设备或系统,其或主要目的是提供以下信息:有关生理或病理状态;或有先天异常;或用于确定安全性以及与可能接受治疗者的相容性;或用于检查治疗措施。
体外诊断试剂的认证主要包括5种不同的方式:
1) Annex III EC符合性自我声明;
2) Annex IV EC符合性声明(全面质量保证体系);
3) Annex V EC型式检查;
4) Annex VI EC产品验证;
5) Annex VII EC符合性声明(生产质量保证体系)。
另:分类属于“Other”的产品在满足IVDD指令要求后,通过Annex III途径,在产品上使用CE标志,无需公告机构参与。
欧盟自由销售证明(Certificate of Free Sale)是指证明产品在特定地域满足相应的法规要求,可以自由销售的文件。
由欧盟国家的主管当局出具的证明企业医疗产品可以在特定区域自由销售的文件,简称为CFS。
欧盟主管当局颁发的自由销售证书,能证明企业生产的产品满足欧盟的法规要求,可以在欧盟市场自由销售。但通常欧盟国家不会要求企业出具CFS,只需CE证书,即可完成清关。
欧盟成员国以外的一些国家,比如埃及、巴西、阿根廷、印度尼西亚、委内瑞拉、沙特等国家会要求企业出示CFS证书。
中国企业申请欧盟自由销售证书CFS 的条件:
(1)指定了欧盟授权代表,签署了书面协议;
(2)产品有合法性的证明,这包括:
a. 如果是I 类的医疗器械,需完成了欧盟注册、CIBG注册;
b. 如果是I*\IIA\IIB\III 类医疗器械,获得了公告机构CE证书。
概括为:核酸检测试剂盒出口欧洲必须办理的项目是:欧代,欧盟注册,注册后证书,欧盟CE IVDD技术文件和DOC.

- CE认证
- 暂无标签