医疗器械产品进入欧洲市场,往往需要通过CE认证。原CE认证遵循的体外诊断医疗器械指令 (in vitro diagnostic medical devices Directive (98/79/EC), 以下简称IVDD) 将于2022年5月26日起被体外诊断医疗器械法规 (in vitro diagnostic medical device Regulation (EU) 2017/746, 以下简称IVDR)所取代。
从IVDD过渡到IVDR,一个显著的变化是更多IVD产品的符合性评估需要公告机构的介入。目前,仅有相当少的高风险产品(大约占到市场上体外诊断试剂的8%)需要按照IVDD的要求提交给公告机构进行审查。施行IVDR后,约有80%的体外诊断试剂需要由公告机构进行审查。
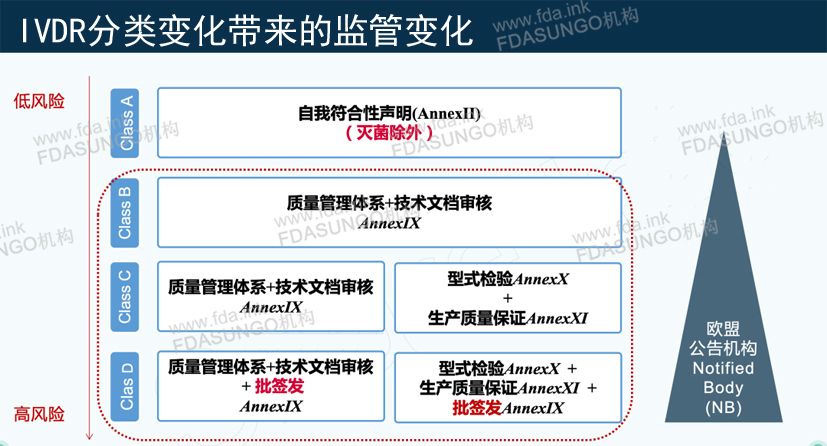
除了Class A类的产品办理CE可以走欧盟注册,欧代,IVDR技术文件合规路径,B类,C类,D类的CE都是需要公告机构做认证,目前取得IVDR的公告机构只有5家,并且这些公告机构因为人员资格等问题,目前还没准备好接IVDR的案件。
无论是新型冠状病毒抗原检测试剂盒还是新型冠状病毒中和检测试剂盒,亦或是新型冠状病毒核酸检测试剂盒,按照98/79/EC指令的分类规则,用于专业人员测试使用的新冠诊断试剂属于List A和List B之外的产品,属于IVDD Ohter类。其CE合规程序是符合性声明,包括企业准备技术文件,签署符合性声明,指定欧盟授权代表,并由欧盟授权代表完成欧盟成员国主管当局注册。
中国NMPA、欧盟IVDR、美国FDA对“体外诊断产品”的定义对比

产品风险等级越高,认证流程越复杂,需要参加的机构越多
(NB、主管当局、欧盟药品管理局、欧盟参考实验室、专家小组)
能够以IVDD Others注册的产品,优先考虑现在直接注册(自我符合性声明):欧盟注册,欧盟授权代表,CE技术文件(含DOC).
对于高风险器械,如HIV或肝炎检测(D类),新要求将从2025年5月开始适用。对于低风险的C类器械,例如某些流感检测,新要求的适用日期将延长至2026年5月,而对于风险更低的医疗器械(B类和A类无菌),适用日期将从2027年5月开始。
特别要注意考虑器械本身在IVDD指令和IVDR法规下的具体分类变化,选用适用自身的合规策略。
FDASUNGO一直倡导制造商基于自身的器械风险类别采取不同的策略。针对产品的特点分别执行了IVDR和IVDD的注册,为客户的产品合规提供最优方案。
已经为国内数百家IVD制造商提供了IVDD和IVDR下的DOC服务,服务内容涵盖上述程序的所有部分,可以帮助IVD制造商及时应对法规变化和新要求。
关于IVD产品我们服务有:欧盟授权代表(荷兰、德国)、欧盟注册、CE技术文件编写、IVDD CE/ IVDR CE认证; 英国代表、UKCA技术文件编写、MHRA注册;ISO13485认证;欧盟自由销售证明CFS。
IVDR分类监管变化
为了制定IVDR下的合规性策略,欧盟委员会建议,制造商应做好差距分析和行动计划,包括以下几个方面:
评估适应新法规给产品带来的影响
核对新的分类规格
确认符合性评估路径
检查和变更已有的技术文件
检查并升级质量管理体系
核对已有临床证据的充分性
风险管理
检查产品说明书、标签
确保上市后监督安排的充分性
准备一份上市后性能跟踪计划
准备应对新的警戒要求
确保溯源义务的实现

- CE认证
- 暂无标签