2024年,康复医疗器械领域的出口额达到了百亿元级别,同比增长12.39%,且连续5年稳步增长。
首先需要明确的是,电动轮椅、电动代步车、拐杖、助行器、沐浴椅、病床等产品在新欧盟医疗器械法规(MDR)下属于CE I类,即低风险产品,管控相对较松。 电动轮椅、电动代步车、拐杖、助行器、沐浴椅、手动病床制造商需要选择欧盟授权代表,完成CE技术文件和欧盟标准检测,并在欧盟主管当局完成注册后,才能在其产品上放置CE标志,以显示其合规性。
FDASUNGO已协助广东、丹阳、浙江、昆山、上海等上百家企业获得CE认证和美国FDA认证,顺利出口欧美市场。
FDASUNGO提供的服务包括CE认证、欧盟授权代表、欧盟注册、美国FDA认证、FDA510K申报、美代、英国UKCA认证、英代、MHRA注册、ISO13485认证、欧盟自由销售证明、瑞士注册、沙特注册、MDSAP认证等。
厂家开始投入欧盟市场,产品进入欧盟必须进行CE认证,以满足欧盟医疗器械法规(MDR)的要求。 以康复器械(如手动轮椅、电动轮椅、手动病床、电动病床、气床垫、担架等)出口欧盟为例,根据医疗器械法规MDR分为I类,不需要公告机构审核,企业可通过自我符合性声明途径进行产品注册,但在注册前需要完成以下步骤:
编制CE技术文件;
完成产品测试(测试标准如电动病床EN 60601-2-52、手动轮椅EN12183、电动轮椅EN12184);
申请SRN号、编制Basic UDI-DI、UDI-DI;
发布DOC符合性声明;
欧盟授权代表;
完成欧盟主管当局注册。
整个周期为3-5周。
FDASUNGO特别提醒广大客户,部分小规模欧盟授权代表服务机构受限于资源投入,在欧盟境内没有合格的PRRC人员,履行职责方面也不能满足当局要求。在当前当局加强监管的情况下,将面临较大的合规风险。
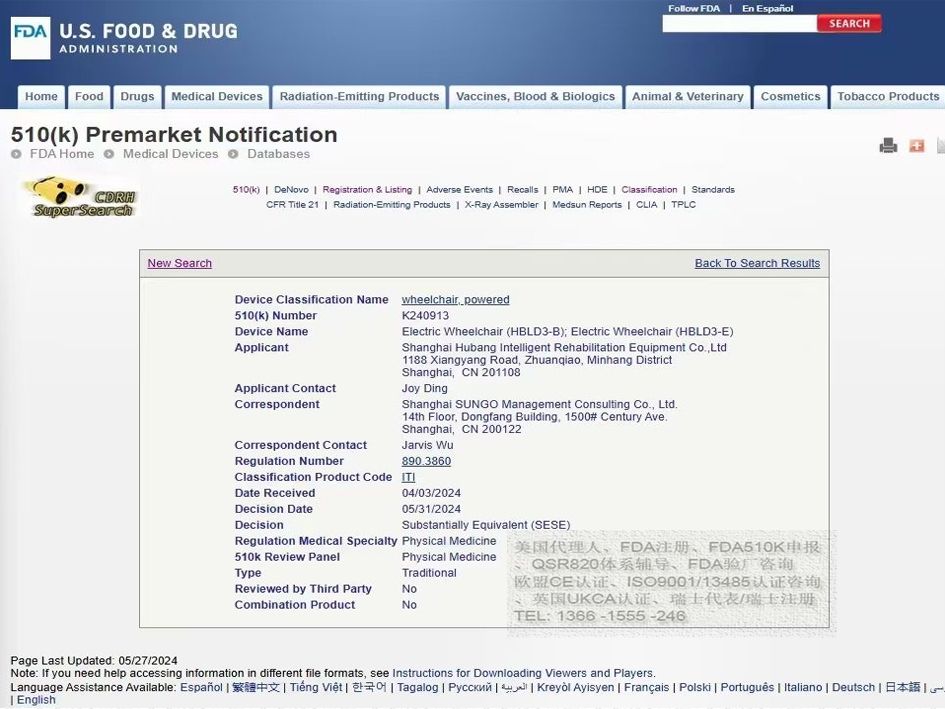
电动轮椅和代步车等产品的美国FDA510K申请流程如下:
电动轮椅检测标准:申请过程中需要提交FDA认可的检测报告,包括ISO7176系统检测、生物相容性检测、安规和电磁兼容等;
电动轮椅510K的关键环节:产品性能、安全测试、控制器和软件验证;
项目周期:FDA的评估到最终批准的时间一般较长,由FDA控制;
通常整个周期在8-12个月左右。
电动轮椅的510K申请流程如下:
按照美国FDA 510K注册标准进行文件分析;
确认现有文件的可用性;
收集并比对市场上已注册的产品信息;
按照美国FDA 510K要求编订产品信息;
根据标准编制510K注册文件;
根据注册文件的评审结果进行修订;
完成公司登记及产品列名注册。
电动轮椅的510K检测标准为ISO7176。选择检测和FDA510K咨询服务至关重要,熟悉检测标准的团队能提高FDA510K申请的成功率,缩短申报时间。



- 美国FDA
- 暂无标签

认证:进军美国市场的核心攻略")