雾化器、尿道扩张器、一次性输尿换导管、气管切开管CE临床评估报告如何编写?
尿道扩张器、一次性输尿换导管、气管切开管属于IIa 产品。医疗器械出口企业在申请CE认证时,不管是I类普通产品还是II/III类高风险产品,都必须要提供第四版临床报告。并且已经拿到CE证书的企业,监督审核也需要提供。
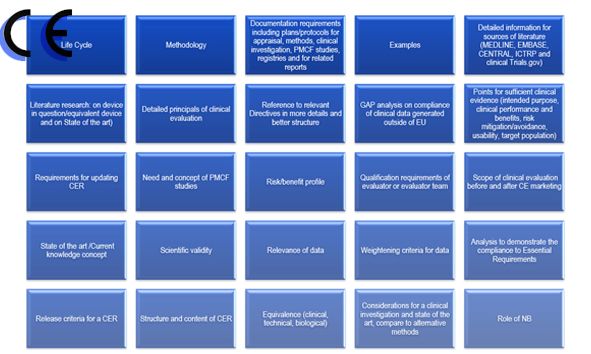
相对于之前的临床报告,第四版的临床报告主要变化体系在:
1.临床报告更新的频率
2.报告编写人和评价人的资质
3.评估报告需要有明确的可测量目标
4.确定技术发展水平
5.数据的科学性和有效性
6.比对器械
7.比对器械的数据获得
8.什么时候需要临床试验
9.售后监督和售后临床跟踪
10.风险—收益。
第四版临床报告编订周期:
通常IIa类产品的编订周期是一个半月左右,IIb类的是2个月,具体周期根据企业的配合度有关。
我司将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发证机构发证公司的全英文临床评估报告。
针对MEDDEV2.7.1 Rev 4,我司可以协助您:
1、协助建立临床评价程序;
2、建立临床评价方案;
3、寻找等同产品,进行等同分析;
4、搜索文献及其他临床数据;
5、临床数据分析;
6、完成临床评价报告;
7、全英文临床评估报告;
8、认证机构审核通过。
FDASUNGO将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发证机构发证公司的全英文临床评估报告。
FDASUNGO可供: CE认证、欧盟授权代表、欧盟注册、CE技术文件编写/编制符合MEDDEV 2.7.1 Rev 4要求的英文临床评估报告、欧盟自由销售明、FDA注册、FDA510K、英国UKCA、瑞士代表/注册、ISO9001/13485认证咨询,合作垂询 1366-1555-246
FDASUNGO临床报告业务优势:
按照第四版临床报告指南的要求,对于临床评估报告的撰写人资格有相应的要求。我司组建了临床评估业务专家技术小组,包括医学博士,国际知名认证机构评审人员,世界500强医疗器械企业质量经理等相关专业人员。目前我司已经交付了近百种产品的临床评估报告,其中包括外科手术导航系统,骨科植入产品等较高风险和复杂程度的产品。例如:一次性无菌注射器带针、一次性无菌输液器带针、电子体温计、医用润滑剂、活组织检查针、输液泵、雾化器、尿道扩张器、一次性输尿换导管、气管切开管、一次性麻醉穿刺针等产品,TUV南德/TUV莱茵/SGS/BSI/KIWA等公告机构要求的欧盟第四版医疗器械临床评价/评估报告,提供编写或更新。
公告机构们于今年也在紧锣密鼓地加强审核!已经有不少企业因此被罚红牌。我司应接不暇, 特此布告医疗器械制造商, 必须尽快做出回应,及时更新您们的临床评价报告(CER) 和质量管理体系(QMS)流程,以符合第四版MEDDEV 2.7/1 rev. 4.0 的要求。
实施计划
对于第四版的执行时间,各家公告机构做法有所不同, 所持的意见也不尽相同。据我们所了解,公告机构的基本思路可以简单归纳为:
-高风险产品和植入器械(例如Class III 和Class IIb), 公告机构期望医疗器械制造商马上执行。
-低风险器械(如 Class IIa, Is, Im), 公告机构会适当放宽期限,有些机构要求2018年内完成更新; 而有些机构则放宽对state of the art 的要求。
应对措施
制造商应该对第四版进行差距分析,从而:
1)对QMS(质量管理体系)的流程进行影响分析;
2)对CER(现有临床评价报告)进行差距分析;
3)对实际更新准备过渡计划(过渡计划应考虑与产品相关风险及证书到期日)。
如何更新CER
-上市后监督信息(PMS & PMCF)
-当前技术水平 (State of the art)
重要信息
PMCF 是强制的,这也是新欧盟法规MDR 突出的重要内容之一。对于下列情形,医疗器械生产商需要做好充分准备:
1)之前的CER (上市临床评价) 走的是等同性路径(特别是高风险产品,如:Class III 和植入器械)
2)产品使用的风险高
3)针对高风险的解剖部位/ 高风险的人群
4)出现了有关安全性和有效性方面的新的信息
5)创新的器械
6)器械的设计适应症和预期用途发生重大的变化。
制造商应该对第四版进行差距分析,从而:
1)对QMS(质量管理体系)的流程进行影响分析;
2)对CER(现有临床评价报告)进行差距分析;
3)对实际更新准备过渡计划(过渡计划应考虑与产品相关风险及证书到期日)。
如何更新CER
-上市后监督信息(PMS & PMCF)
-当前技术水平 (State of the art)
所以,将要申请或者已经拿到了TUV莱茵、TUV南德、SGS或其他公告机构CE证书的企业,一定要高度关注。

- MDR认证
- 暂无标签
