一、什么是UDI?
唯一器械标识(Unique Device Identification,缩写UDI)是美国FDA建立的”特殊医疗器械的识别系统”,是对医疗器械在其整个生命周期赋予的身份标识,是其在产品供应链中的唯一“身份证”。主要是一个由数字或字母组成的编码。由器械识别码(DI)和生产识别码(PI)组成。器械标识DI属于静态信息,它是医疗器械产品在供应链中的身份标识,可作为进入数据库查询该产品追溯基本信息的“关键字”;而生产标识PI属于动态信息,它包括医疗器械产品的序列号、批号、生产日期和有效期等,是医疗器械产品的动态附加信息,它与DI联合使用,才能指向特定的医疗器械产品。
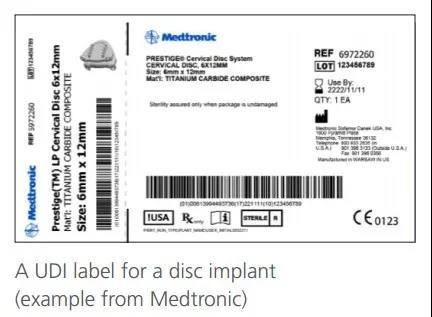
二、UDI代码的结构和编制方法
UDI是一个由数字或字母组成的编码。由器械识别码(DI)和生产识别码(PI)组成。

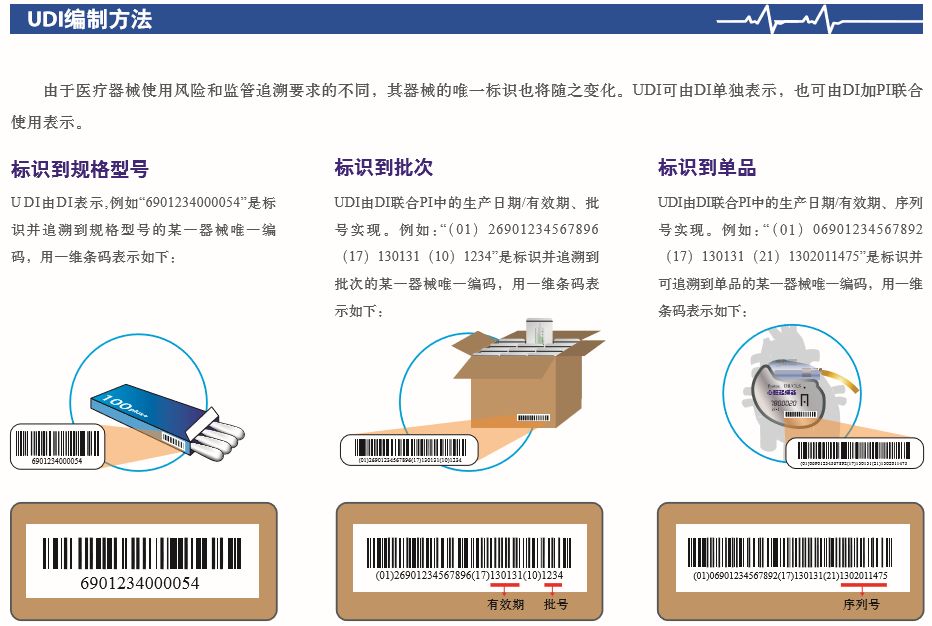
三、 UDI数据载体

四、 实施时间要求
|
强制实施日期 |
执行要求 |
|
2014 年 9 月 24 日开始 |
- 所有 FDA Class III 第三类医疗器械;FDA PHS Act 法案管制的器械,都必须在器械的标签和包装上标注 FDA UDI 信息,采用 FDA 规定的日期格式并及时报送 FDA GUDID 数据库; - 如果特定企业需要申请延期执行 UDI,务必在 2014 年 6 月 23 日向 FDA 递交申请; - FDA Class III Stand-alone Software 被 FDA 作为第三类别管制的独立软件必须提供 UDI 信息 |
|
2015 年 9 月 25 日开始 |
- Implantable, life-supporting, and life-sustaining devices 植入式医疗器械,用于生命支持和 维持的医疗器械的标签和包装都必须标注 UDI 信息,并采用 FDA 规定的日期格式; - 如果上述器械为重复使用器械并在使用前可被再处理,那么必须进行永久性 UDI 标识; - 用于生命支持或维持器械的独立软件必须标识 UDI; - 上述器械 UDI 数据必须报送 FDA GUDID 数据库; |
|
2016 年 9 月 24 日开始 |
- UDI 管制的 Class III 可重复使用并且在使用前可以被再处理的医疗器械都需要进行永久 性 UDI 标识; - FDA Class II 二类器械的产品标签和包装必须标识 UDI,并采用 FDA 规定的日期格式; FDA Class II Stand-alone Software 被 FDA 作为第二类别管制的独立软件必须提供 UDI 信息; - FDA Class II 二类器械的 UDI 数据以及产品关键数据必须报送 FDA GUDID 数据库; |
|
2018 年 9 月 24 日开始 |
- FDA Class II 可重复使用的并且使用前可以被再处理的 FDA 管制的二类器械必须进行永久性 UDI 标识; - FDA Class I 一类医疗器械和未被划分级别 Class I, II 或 III 的器械都必须标识 UDI;所有这些器械,包含豁免 UDI 的器械,日期标注都必须符合 UDI 法规; -上述器械的 UDI 信息必须及时报送 FDA GUDID 数据库; - FDA Class I 一类的 Stand-alone software 独立软件必须提供 UDI; |
|
2020 年 9 月 24 日开始 |
- 所有 FDA Class I 一类器械和未被分类为 Class I, II, III 的器械,如果可以重复使用或使用前可以被再处理,都必须在产品上进行永久性 UDI 标注; |
|
注意:在强制日期前生产以及贴好标签的器械, 可以不需要执行 FDA UDI 要求,豁免只能在生效强制日期后三年内采纳。 超过三年一律要执行。
I类的器械目前也可以按照UDI的规定开展工作。但并不是以2018年9月24日作为截止时间!! |
|
五、UDI申请流程
第一步:申请和设计UDI
1. 向签发机构申请产品编码(DI)
2. 编制UDI (由DI+PI组成,DI向签发机构申请,PI厂商按照编码规则自行编制)
3. UDI 运行(将UDI加印在医疗器械上)
第二步:申报和维护UDI
4. 收集GUDID 需要的信息(获取邓白氏码、获取GMDN code)
5. 申请GUDID账号
6. 数据提交GUDID
六、 我们的服务
|
UDI项目辅导 |
1.帮助企业进行项目规划 |
|
2.向签发机构申请DI |
|
|
3.帮助企业编制UDI,并写入程序文件 |
|
|
4.帮助企业建立贴标的标准操作流程 |
|
|
5.收集相关数据,为UGDID申请做准备 |
|
|
UGDID申报 |
1.作为企业的UDI法规事务联络人 |
|
2.UGDID账号申请 |
|
|
3.UGDID数据提交 |
|
|
4.UGDID数据更新与维护 |